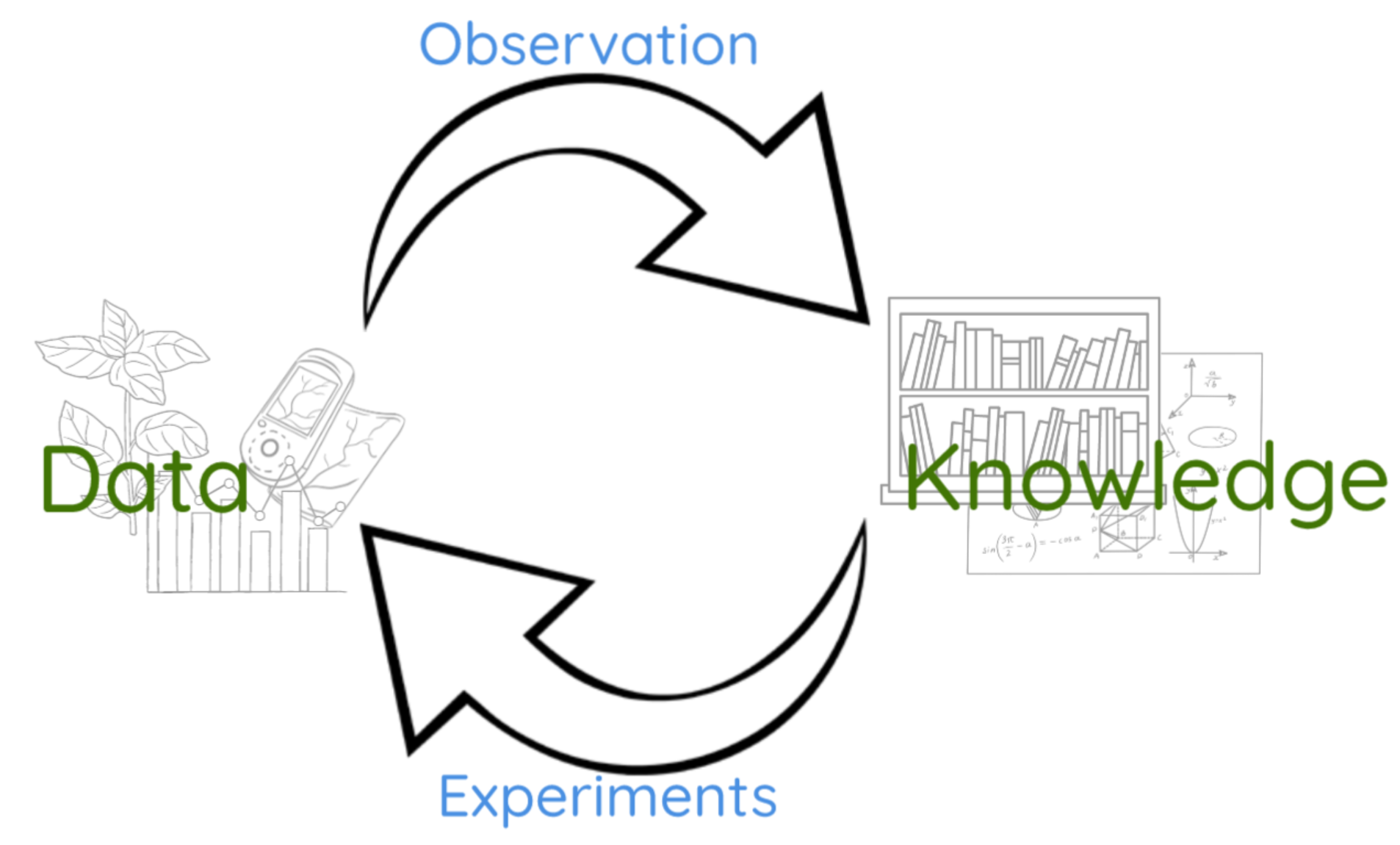
The modern western tradition of scientific discovery was instigated by Francis Bacon in 1620, and considers the process by which knowledge is obtained. Data, which is measured in the natural world, is converted into knowledge by a process of observation. However not to be forgotten, especially in the big data era, that data is measured for a reason. Data is measured via the so-called process of experimentation, where our knowledge if the real world is used to design which data is captured.
Anatomy of Machine Learning Enabled Scientific Discovery
Taking the lens of the scientific method, we can unpack the different kinds of machine learning tasks that are often lumped together in the popular media as “artificial intelligence”. We decompose the scientific method into four phases: representation, prediction, decision, and governance.
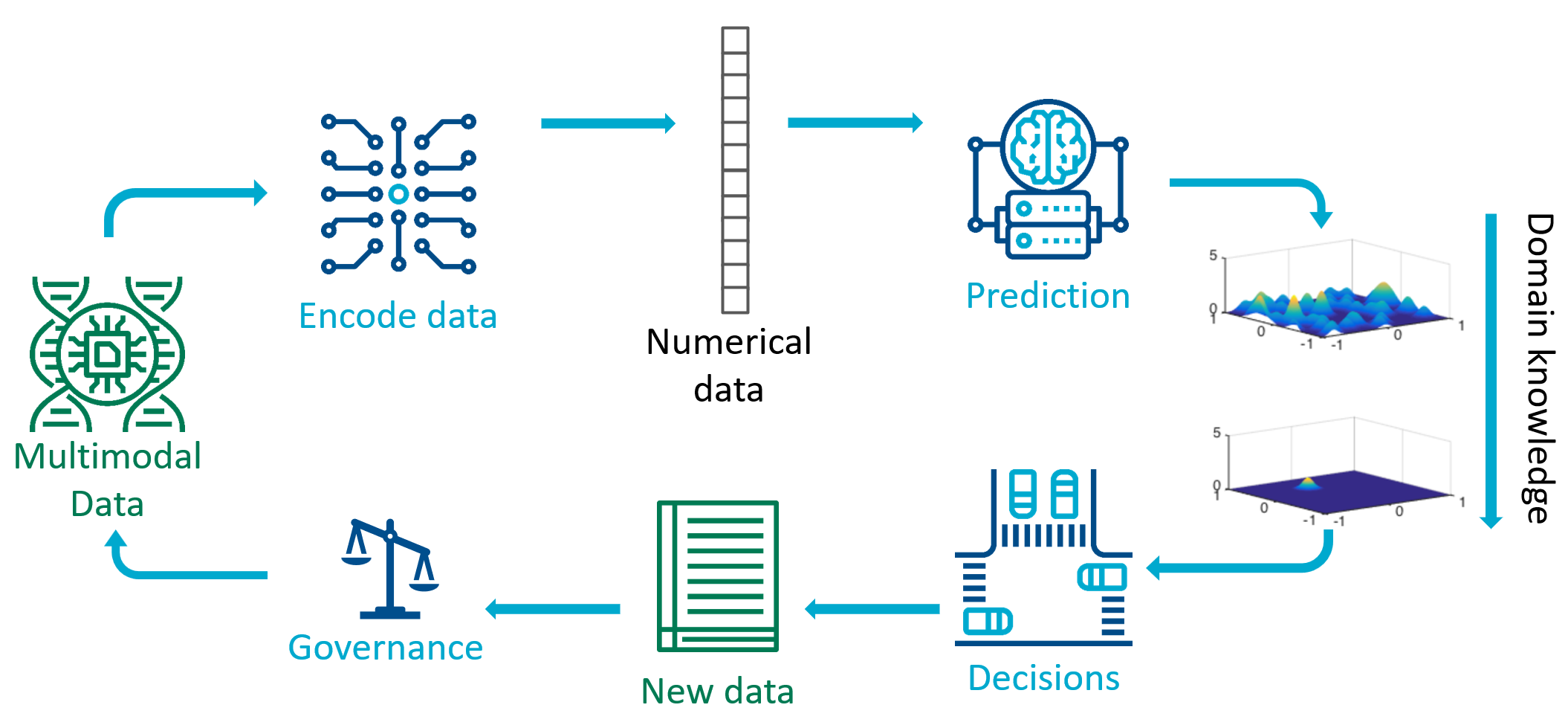
Data measured from sensors such as cameras, DNA sequencers, and web databases, are not immediately amenable to machine learning predictions. A first step is to identify a numerical representation, that is to convert the data from sensors into a suitable mathematical representation as vectors on a computer. This step has been named: finding an embedding, representation learning, or estimating a metric. This challenging step has seen many success stories in recent decades, where we have found good representations for parts of words of natural language, and patches of images, enabling efficient ways to manipulate data in natural language processing and computer vision.
Once in a suitable vector representation, methods for prediction can be brought to bear. Predictive machine learning methods, such as random forest, support vector machines, and deep neural networks, tends to be the main focus of undergraduate courses. The goal of this step is to take a vector representation of data, and compute a transformation that results in a more human accessible numerical representation. For example to take a vector representation of an image, and compute/predict that the image contains an object of class “57”, which has been allocated to mean “cat”. Some predictive machine learning methods additionally include predictive uncertainties or confidence scores. For example the predictor may output “cat” with probability 0.8, and “possum” with probability 0.2.
While domain knowledge is critical for all phases of the scientific discovery process, when using machine learning approaches it is convenient to inject domain knowledge between the prediction and decision phases. Domain knowledge could be used to constrain the kinds of predictions that are allowed, for example ensuring that conservation of mass is respected when predicting physical quantities. Predictive models have had great success in machine learning, including recognising written text from images, classifying objects, and predicting 3D protein structures from the amino acid sequence.
Given these predictions and their associated uncertainties, the next phase is to make decisions. In the process of scientific discovery, the scientist makes a decision about which experiment to perform next. The scientist considers the infinite set of all possible experiments, and makes a decision which measurement to take and how to perform the measurement. There are several different machine learning approaches for decision making, including active learning, Bayesian optimisation, and reinforcement learning. As we deploy machine learning algorithms more broadly in society, we need to consider impacts of automated decision making and create sociotechnical governance structures. One key aspect related to the scientific method is the question of whether the data we measure is representative of the population under study. In classical statistics, experimental design methods provide ways to ensure coverage of the population, but there are many modern applications of decision making in society that require further research in methods for ensuring fairness, accountability, and transparency.
Illustration using Engineering Biology
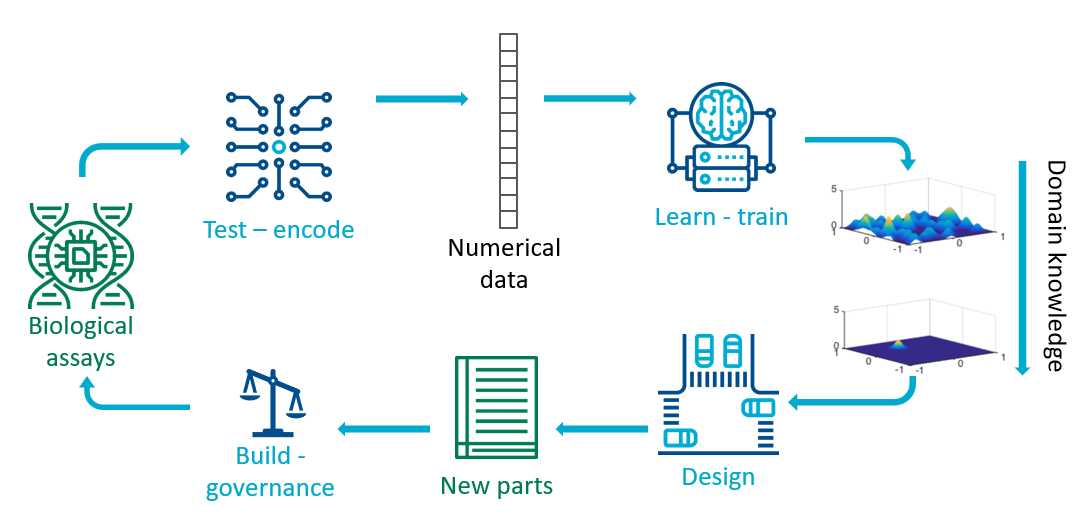
The research area of synthetic biology frames the scientific method as the Design-Build-Test-Learn (DBTL) cycle. Using the perspective presented here, for a particular organism or biological part, the Test part of the cycle uses a variety of assays (for example measurement of omics) to obtain data that a particular system performs as desired. As in the chatbot example, the outputs of assays are converted into a numerical representation. For example if the assays are DNA/RNA sequences, they are split into substrings called k-mers which are the tokens which are then transformed by deep learning methods. The coincidentially named Learn step is the step where we train a machine learning predictive model. Given trained predictive models (and their associated uncertainties), we can use experimental design approaches to Design new biological parts. The overarching goal of synthetic biology is to engineer useful biological parts and to find ways to combine them. Once the parts have been designed, complex protocols still need to be developed to Build the part. These protocols would in future build on current regulations on safety and ethics of genetic modifications to provide governance structures for engineering biology.
Decisions - Where to Measure?
A critical research question that is highly important for many application of machine learning in scientific discovery is to identify “good” designs. In this section we unpack the component named Decisions in the anatomy.
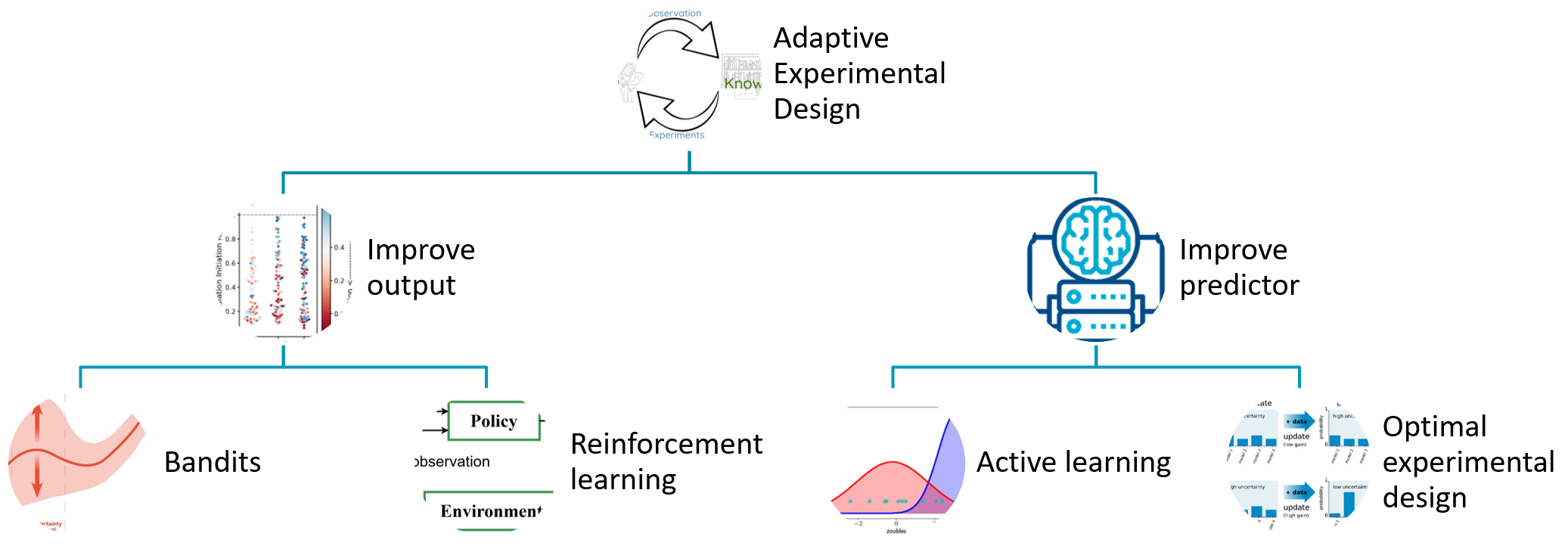
In scientific discovery, the decision facing a scientist is to design experiments that measure future data. Good designs may be to improve the model, as in the example of chatbots. The developer of chatbots would be interested to present sentences to a human annotator whose annotation would most significantly improve the generation of future sentences (improve the predictor). In contrast the goal of good designs may be to improve the measured yield, as in the example of synthetic biology. The life scientist may be interested to engineer biological parts that improve the production of a particular protein of interest, for example an enzyme that digests plastic faster than before.